
Chapter 13: Introduction to Bioinformatics
Ursula Frei; Walter Suza; Thomas Lübberstedt; and Madan Bhattacharyya
A biological sequence database is a collection of molecular data organized in a manner that allows easy access, management, and update of the data. Biological sequence databases serve an important role of providing access to sequence information to the research community. The databases contain molecular information of multiple organisms and are constantly being updated and re-designed to allow more robust data query and analysis. Examples of biological databases include European Molecular Biology Laboratory (EMBL), GenBank, the National Center for Biotechnology Information (NCBI), and the DNA Databank of Japan (DDBJ). Every sequence submitted to the database has a unique number assigned to it, called the Accession number. Even if the same gene has been submitted several times by different investigators each will have a different accession number. The chapter includes practical examples of using database tools. It is recommended that you use “try this” questions to become familiar with sequence databases.
- Familiarize with some of the most commonly used databases in molecular plant breeding
- Learn the tools for accessing and manipulating biological databases
- Develop proficiency in the use of biological databases
Database Types
Databases can be classified in to primary (archival), secondary (curated), and composite databases.
- A primary database (e.g. EMBL/DDBJ/GenBank for nucleic acids) contains information of the sequence or structure alone, for example, DNA, RNA, or protein sequences.
- A secondary database (e.g. eMOTIF at Stanford University, PROSITE of Swiss Institute of Bioinformatics) contains information derived from the primary databases and represent sequences that are consensus of a population, for example, conserved features and motifs of a sequence.
- A composite database contains a variety of different primary databases and provides multiple options for database search (e.g. NCBI, MaizeGDB). New tools are continuously developed to make both submission and access to sequence databases more efficient.
Access and Use of Sequence Databases
Once a new sequence has been determined a common step in its analysis is to compare the sequence with related genes that have already been sequenced, often from other organisms. A few things to keep in mind about database searches and sequence databases in general:
- Do not assume that if a sequence is in the database it must be correct. Databases are full of errors!
- Similarity with a known protein or gene does not necessarily mean the query is the same gene as the one it has similarity with.
- Two nucleotide sequences may have low similarity yet code for proteins that are functionally related.
- Protein sequences may also have low similarity yet still be functionally or structurally related.
About NCBI
NCBI was created in 1988 as a division of the US National Library of Medicine at the National Institute of Health. The role of NCBI is to create automated systems for storing and analyzing sequence information.
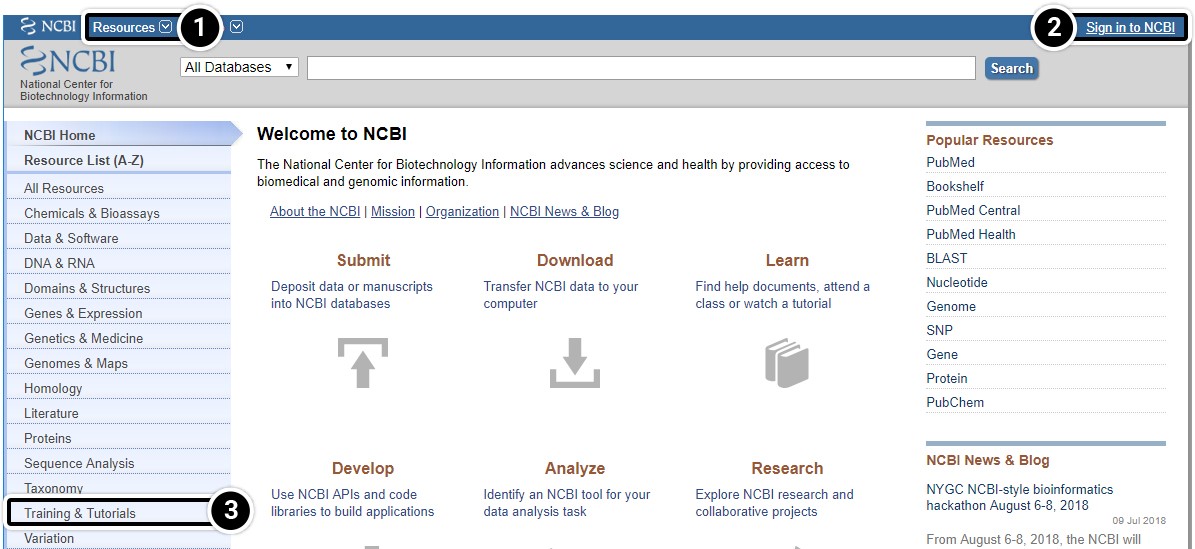
- To access various resources available through NCBI select Resources.
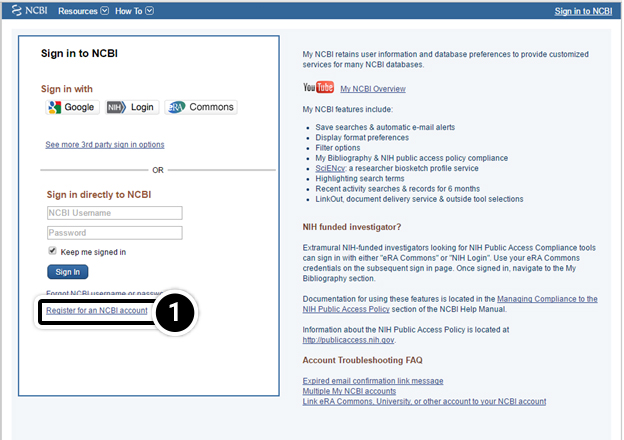
- We recommend that you set up an account with NCBI to allow you the option of saving your results. Click the Sign in link to do so
- Video tutorials are available under the Training & Tutorials link to enhance learning.
Sign Up for NCBI
1. Click Register to set up a new NCBI account.
NCBI Training
NCBI was created in 1988 as a division of the US National Library of Medicine at the National Institute of Health. The role of NCBI is to create automated system for storing and analyzing sequence information.
Information Retrieval from NCBI
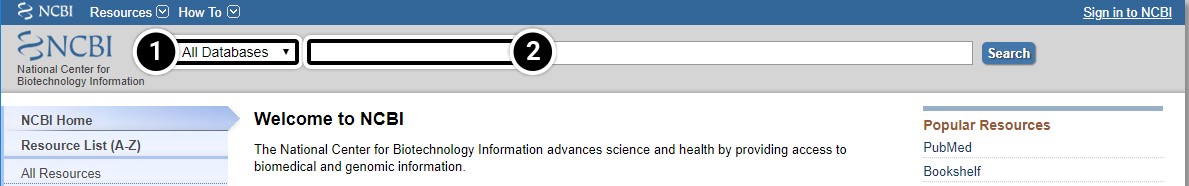
One of the most widely used interfaces for the retrieval of sequence information from biological databases is the NCBI Entrez system. Entrez relies on preexisting, logical relationships between the individual sequences (data points) available in various public databases.
- Searching all databases is often a good starting point to get an overview of the state of your research field.
- Searches are based on keywords.
Searching NCBI by Keywords
Searches can be restricted to a single database or expanded to include all other databases. The simplest way to query is through the use of individual search terms, coupled by Boolean operators such as AND, OR, or NOT. A Boolean operator is a variable that can have only a true or false value.
- Select individual databases, or search them all.
- AND: To ‘AND’ two search terms together instructs Entrez to find all documents that contain BOTH terms
OR: To ‘OR’ two search terms together instructs Entrez to find all documents that contain EITHER term.
NOT: To ‘NOT’ two search terms together instructs Entrez to find all documents that contain search term 1 BUT NOT search term 2.
This activity consists of the following steps:
Step 1: Compare the sequences
Compare the sequences for the adh1 gene in maize and sorghum. Navigate to the NCBI site.
- Enter adh1 in the “search across databases” window. How many adh1 candidates did your search find?
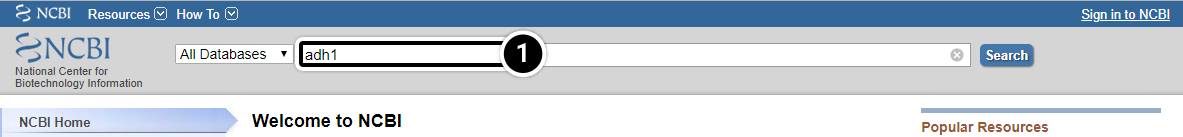
Step 2: Results of a search
Results of a search for “adh1” across all databases:
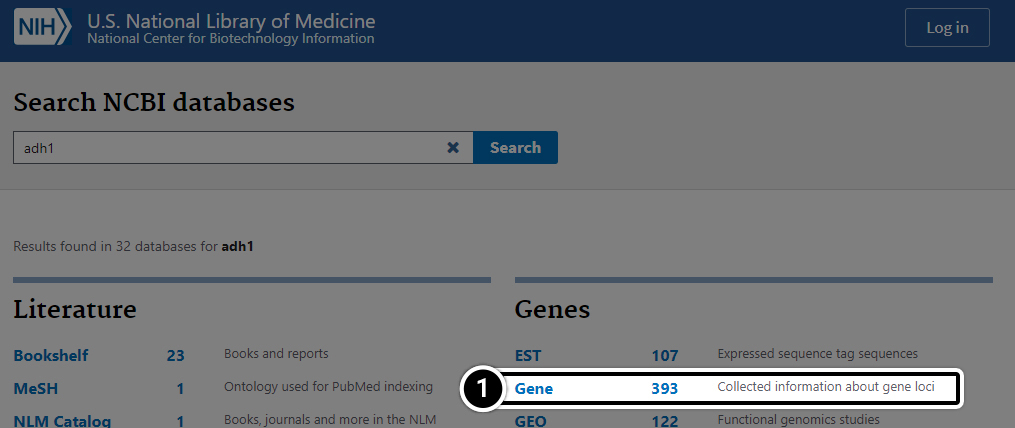
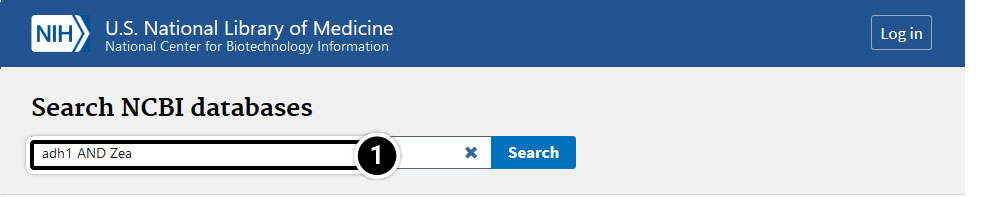
Step 3: Compare the results
Compare the sequences for the adh1 gene in maize and sorghum:
- Enter adh1 AND Zea in the search window.
Compare the results in the Gene category.
- Boolean operators can be used to restrict a search and allow users to obtain specific information about their organism of interest.
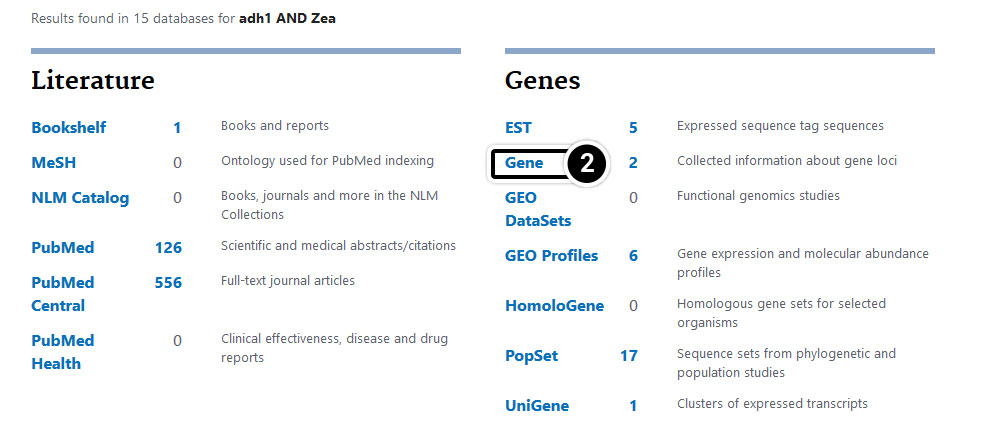
Step 4: Operators
Now try these operators.
- Enter adh1 AND Zea[orgn] OR Sorghum[orgn] in the search window.
- Results
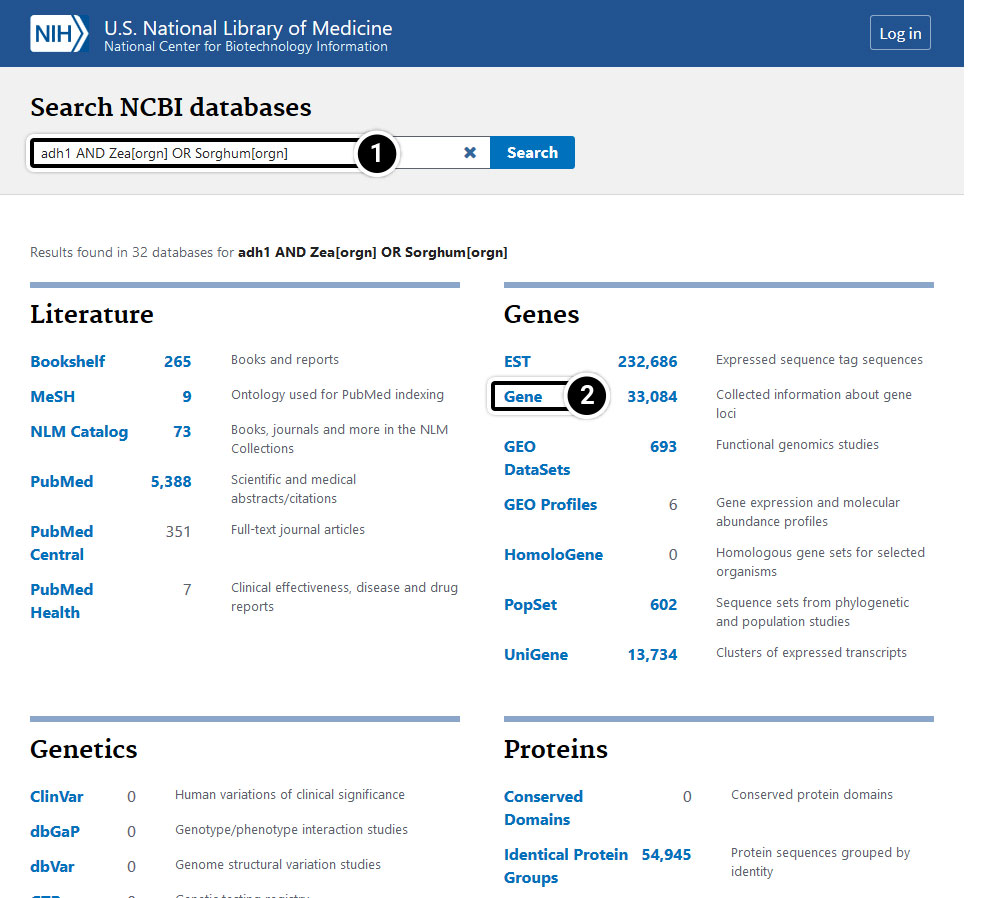
Step 5: Operators
Now try these operators.
- Enter adh1 AND Zea[orgn] OR Sorghum[orgn] in the search window.
What stands out when you compare results when using the search terms “adh1 AND Zea[orgn] OR Sorghum[orgn]” and “adh1 AND (Zea[orgn] OR Sorghum[orgn])? Can you identify any differences among the results obtained from the following sets of search terms?
- “adh1 AND Zea[orgn]” and “adh1 AND Zea[orgn] OR Sorghum[orgn]”
- “adh1 AND Zea[orgn]” and “adh1 AND (Zea[orgn] OR Sorghum[orgn])“
- Enter adh1 AND (Zea[orgn] OR Sorghum[orgn]) in the search window.
- Compare the results in the Gene category.
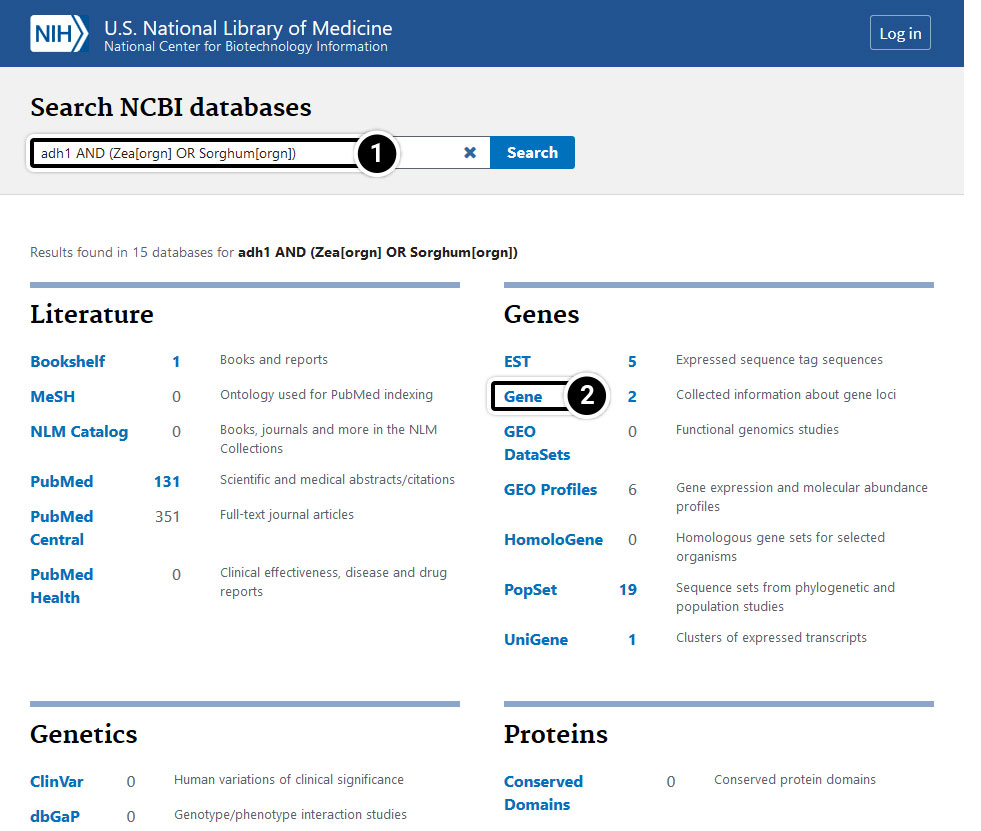
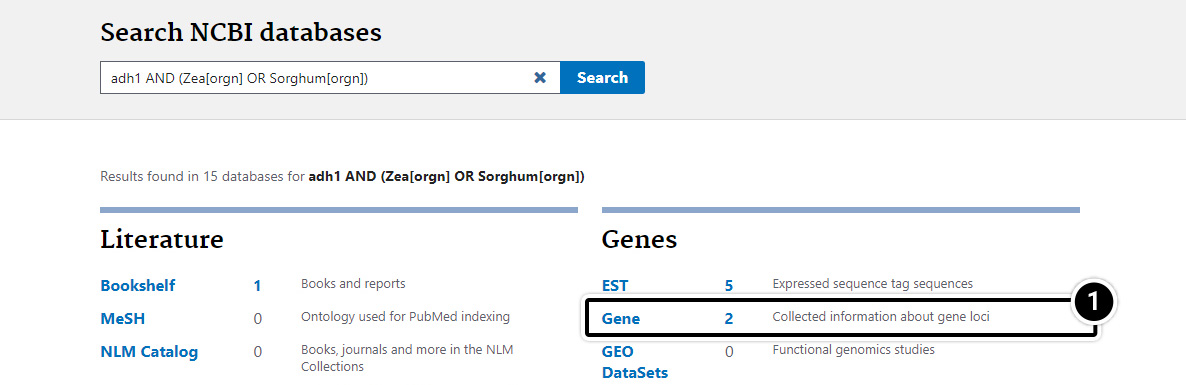
Step 6: Gene-centered Info
- Click on “Gene” to get gene-centered information on the output in the last screen results (also shown here).
- Click on the first “adh1″.
- Review the output window.
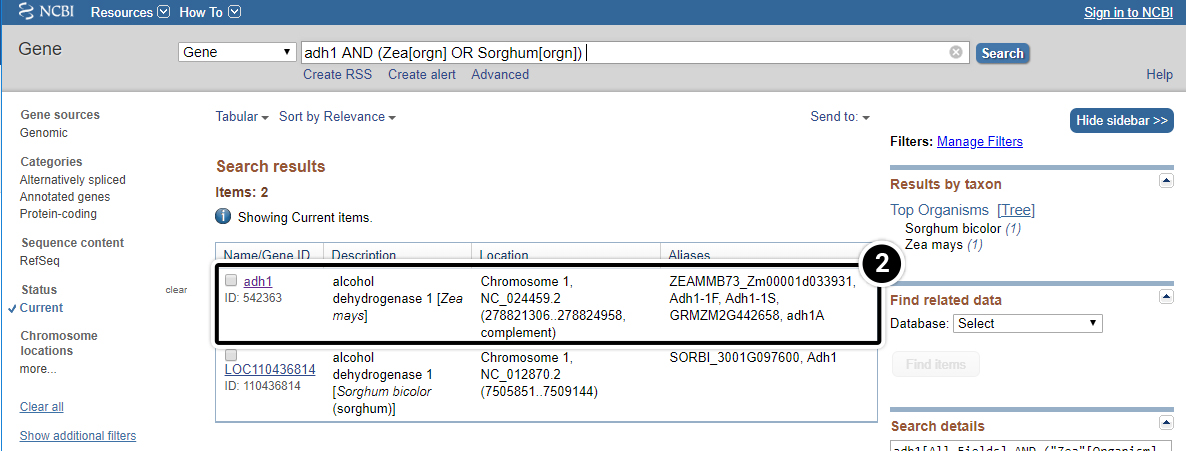

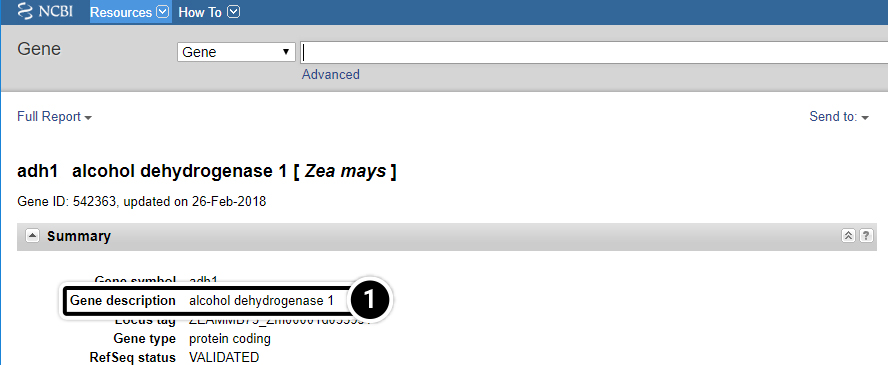
Step 7: adh1
What is the function of adh1?
- Answer found below.
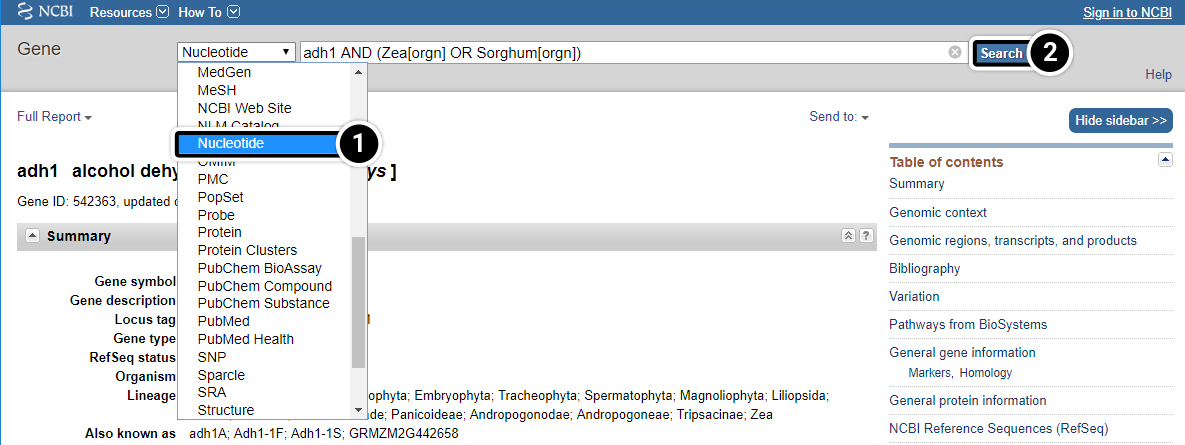
Step 8: Nucleotide results
Let’s examine the Nucleotide results.
- Click this pull-down menu for more information about this gene and select Nucleotide.
- Click Search. Your search will result in 100s of hits.
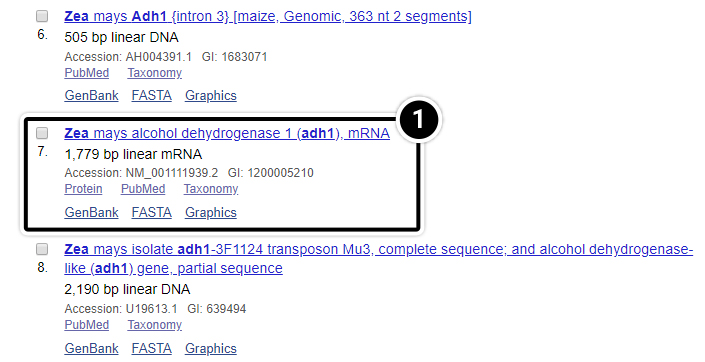
Step 9: Nucleotide results
Let’s examine the Nucleotide results.
- After selecting the nucleotide option as in previous screen, click on the adh1 mRNA as indicated. You may have to scroll down to find it.
Let’s examine the Nucleotide results.
- Click the FASTA link
- Reference sequences are accessed through GenBank to provide non-redundant curated data derived from experimental knowledge of known genes.
Additional information about RefSeq can be found here.
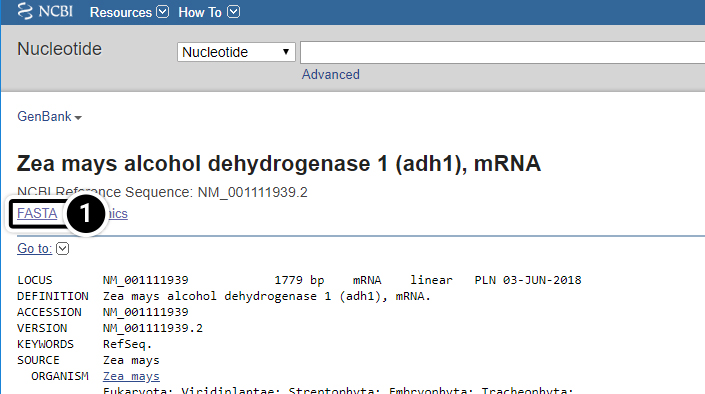
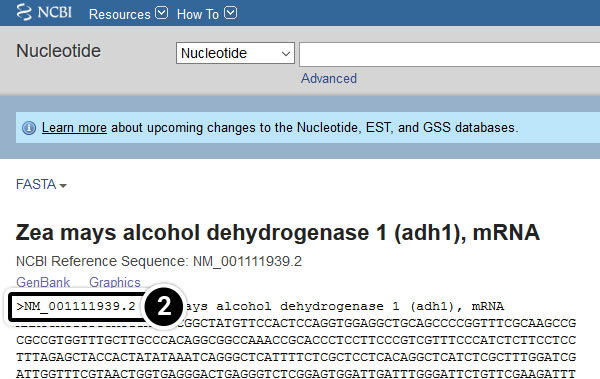

After clicking the FASTA link, what kind of information do you get?
Does the entire mRNA sequence for adh1 you obtained code for a protein product? If not, how would you identify the coding sequence?
NCBI BLAST
NCBI Basic Local Alignment Search Tool (BLAST)
Not only keywords can be used to search sequence databases. Sequences can also be used to perform a BLAST search, making BLAST probably the most important tool in any sequence database. BLAST allows the comparison of sequence data using an algorithm developed by Altschul et al. (1990). The algorithm attempts to detect high-scoring segment pairs, which are pairs of sequences that can be aligned with one another and, when aligned, meet certain scoring and statistical criteria.
BLAST Interface
On the BLAST Interface, the user can restrict searches to a specific species and to the assembled reference sequences for that species. For a plant researcher, it may not be necessary to restrict a search except for those working with rice and Arabidopsis. For all other plant species reference sequences are not fully developed.
BLAST Features
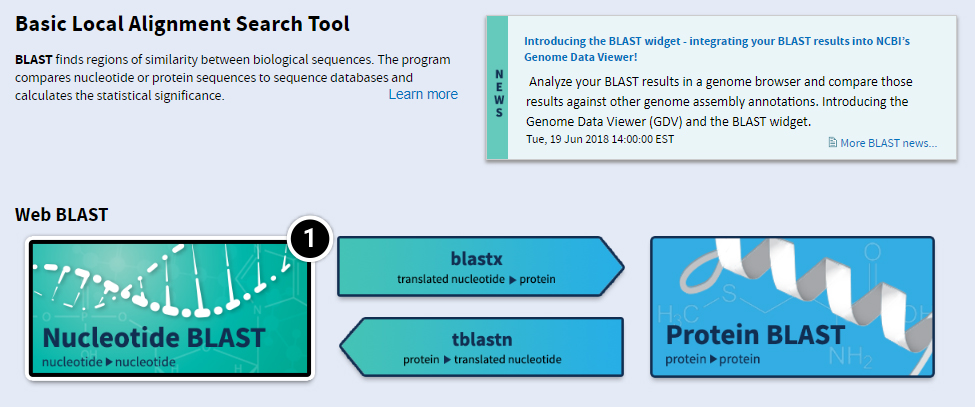
- Basic BLAST features include blastn, blastp, blastx, tblastn, and tblastx.
- Specialized features include “Global Align” for sequence alignment.

Try This: Using NCBI BLAST
- Within the Basic BLAST window, click on Nucleotide BLAST. A new window appears asking you to setup your search options.
- This is where your query sequence will go.
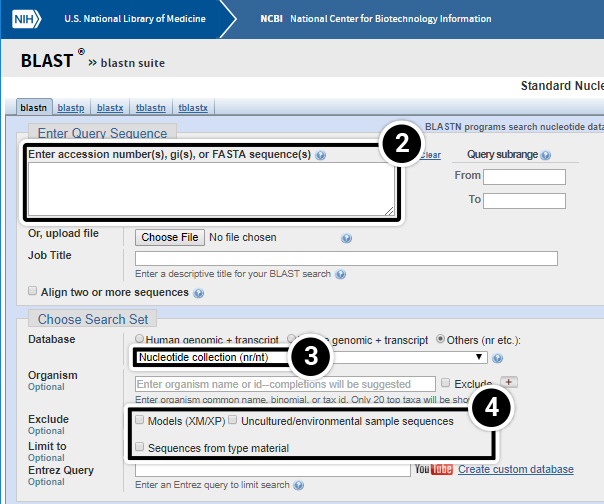
- This selects the Database you want to search.
- Other parameters you may want to set different from the standard settings.
You have various options of entering your query sequence: copy and paste or uploading a saved sequence from your computer.
Your query sequence has to be annotated in FASTA format. FASTA is a text-based format consisting of a definition line followed by the sequence data in single letter code. The definition line starts with the character “>”, followed by a sequence name, and ends with a return or newline. Everything that follows until the next “>” will be considered as the sequence data. It is possible to save multiple sequences in one FASTA file.
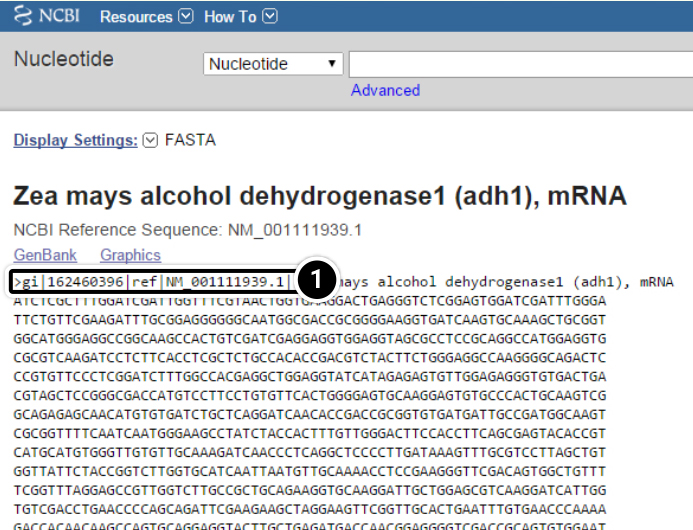
- In the screenshot below,
Definition line starts with “>” character,
gi stands for GenBank identification, followed by GenBank ID number,
ref stands for reference sequence, followed by the accession number.
Both GenBank ID and reference sequence numbers can be used to enter a query sequence into BLAST.
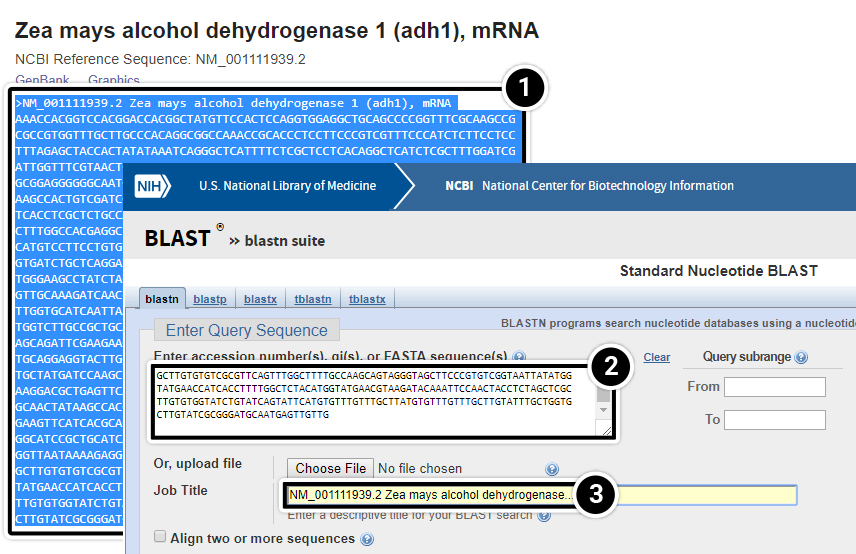
You may enter your query (adh1) as a sequence in FASTA format.
- To do that, copy the entire adh1 sequence
- Paste it in the Enter accession numbers(s), gi(s), or FASTA sequence(s) window.
- Note that the Job Title filled automatically.
Try This: Using NCBI BLAST
- Alternatively, you can query your sequence using the Run BLAST command. Click “Run BLAST” to query the sequence from the FASTA display screen.

Try This: Using NCBI BLAST
Clicking on the Run BLAST command will lead you to this window.
- Accession number of adh1 will automatically fill in
- Job Title should automatically fill in, if it does not you can click in the Job Title field and it should appear automatically.
- Optimize your search to megablast to identify highly similar sequences.
- Finally, select the BLAST button.
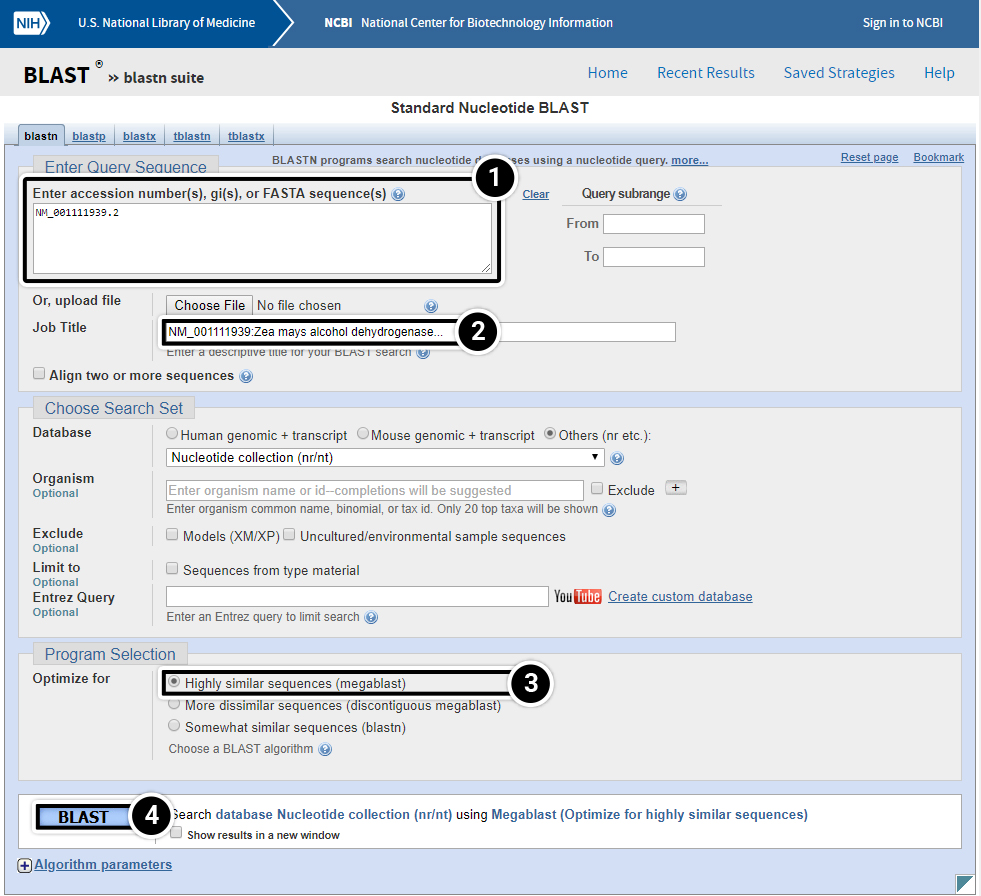
Try This: Using NCBI BLAST
- Graphic Summary: BLAST results that are summarized in a graphic form.
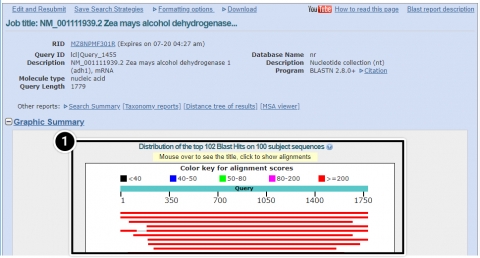
Try This: Using NCBI BLAST
- Alignments: BLAST results that contain sequence alignment information.
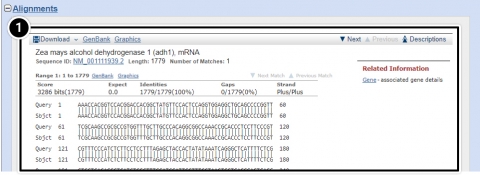
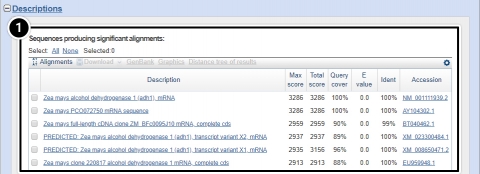
Try This: Using NCBI BLAST
- Descriptions: Accession number and source organism information is provided for sequences producing high alignment scores.
Try This: Using NCBI BLAST
Step 4: Locating adh1 on a chromosome
- From the NCBI home page, select Genome.
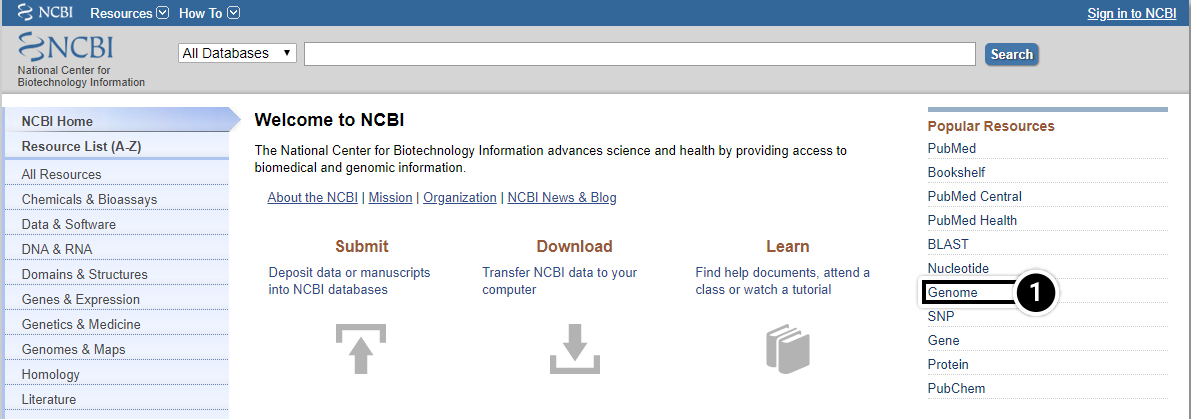
Try This: Using NCBI BLAST
Locating adh1 on a chromosome
- From the genome page, select Genome Data Viewer (previously known as Map Viewer).
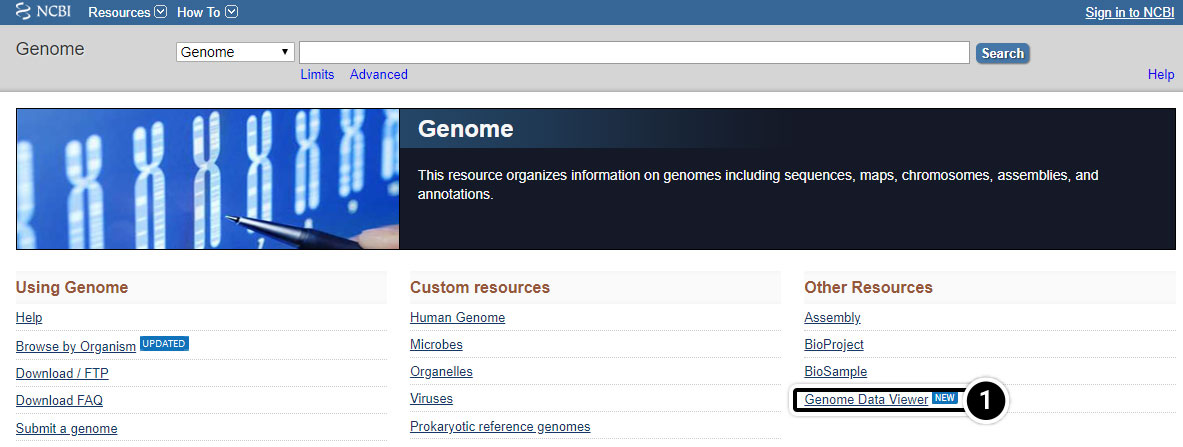
Try This: Using NCBI BLAST
Locating adh1 on a chromosome
- Within Genome Data Viewer home you can select your organism or species.
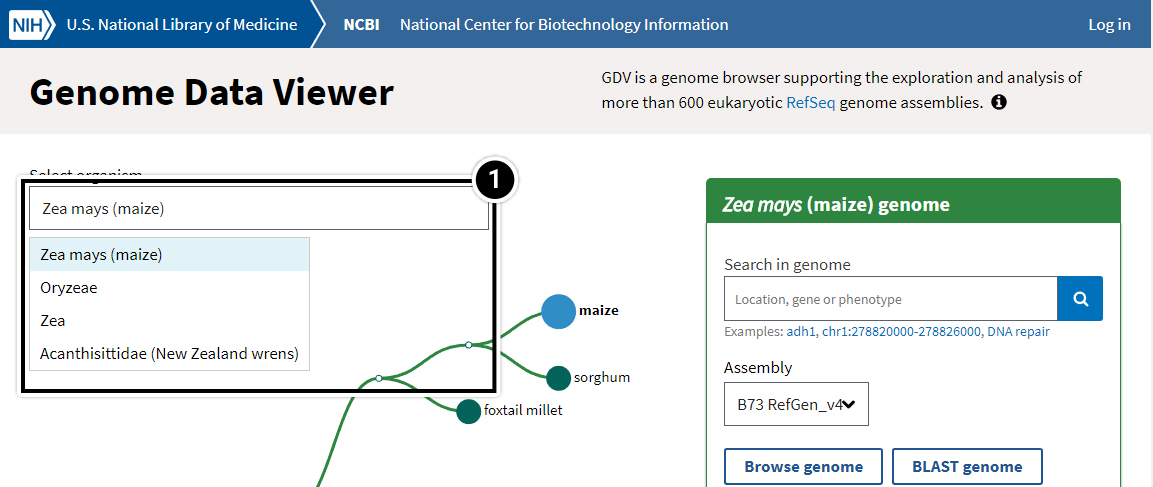
Try This: Using NCBI BLAST
Locating adh1 on a chromosome
- Try searching the Zea mays genome for the adh1 gene.
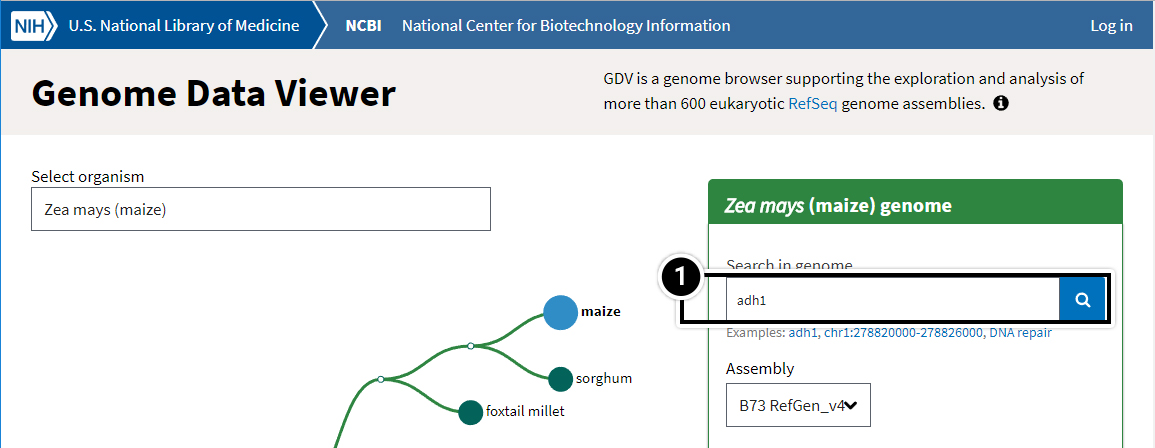
Try This: Using NCBI BLAST
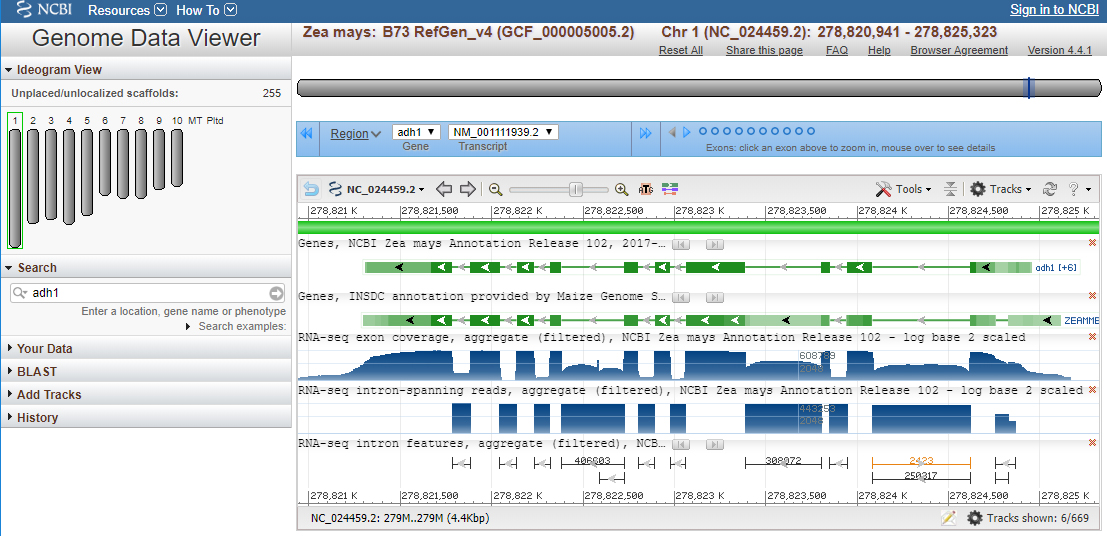
The NCBI Map View search for adh1 on the maize genome produces these results. “Ideogram view” highlights chromosome 1 to show that the adh1 gene is located on chromosome 1.
Plant Species Sequence Databases
The advent of genomics has resulted in a number of plant species specific sequence databases. For this lesson, Maize Genetics and Genomics Database (MaizeGDB) will be the focus.

MaizeGDB
MaizeGDB was first released in 1991 (as MaizeDB) and has transitioned from a focus on curation of genetic maps and stocks to the handling of reference maize genome sequence, multiple maize genomes, and sequence-based gene expression data. MaizeGDB relies on the research community for data and on expertise distributed across the USA. We recommend the use of an internet browser other than Internet Explorer (e.g. Google Chrome) to access the MaizeGDB site.
- Tutorials are available under the About menu under “Outreach.”

MaizeGDB: Tutorials
Useful MaizeGDB tutorials are available to help the user become familiar with the tool.
Step 1: Perform a Basic Search
Similar to NCBI, the MaizeGDB is a composite database allowing you to search broadly among databases or to restrict your query to a single database.
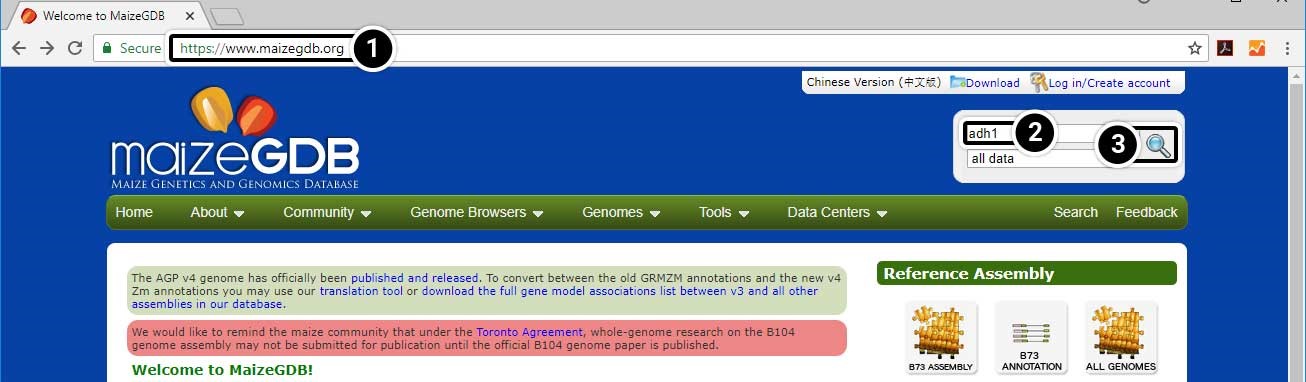
- Open your web browser and go to https://www.maizegdb.org.
- Enter adh1 into the search box.
- Press Enter or click the Search icon to search within all available data.
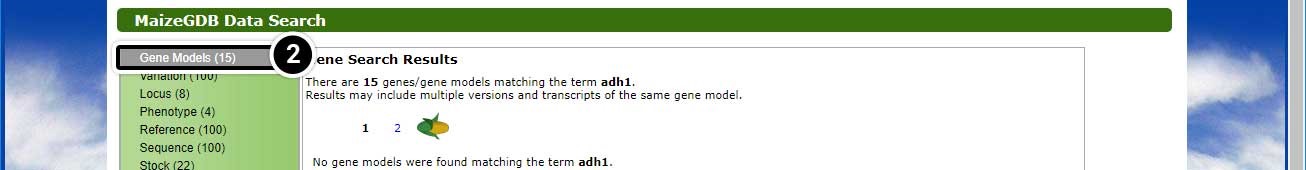
Step 2: Explore the Search Results
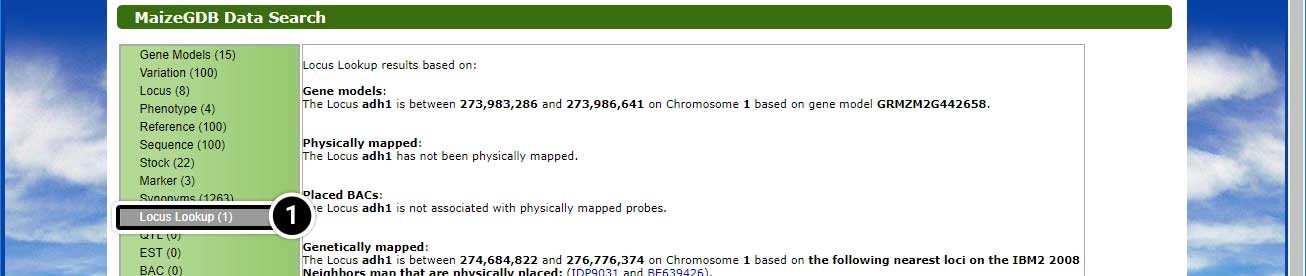
This search will lead you to a window containing various options.
- Click on Locus Lookup (1) in the left-hand menu.
- Click on Gene Models (15) in the left-hand menu.
Explore the other data available to you by clicking the links in the green box. Click the image below to see a larger version.
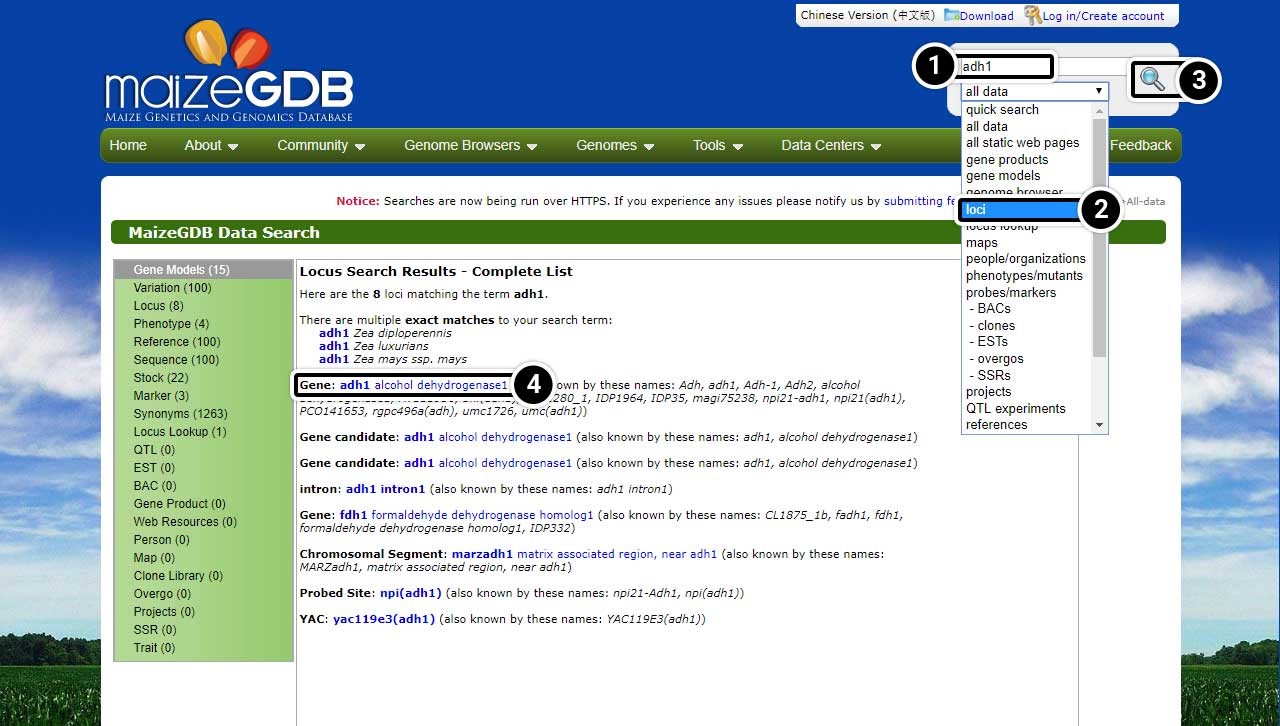
Step 3: Access the Genome Browser
Access the genome browser to obtain information about maize adh1.
- Type adh1 into the Search bar.
- Select loci from the Search options.
- Click the Search icon.
This time when the results load, you will see only the loci associated with adh1.
- Click the link for adh1 alcohol dehydrogenase1 to take a closer look at the gene.
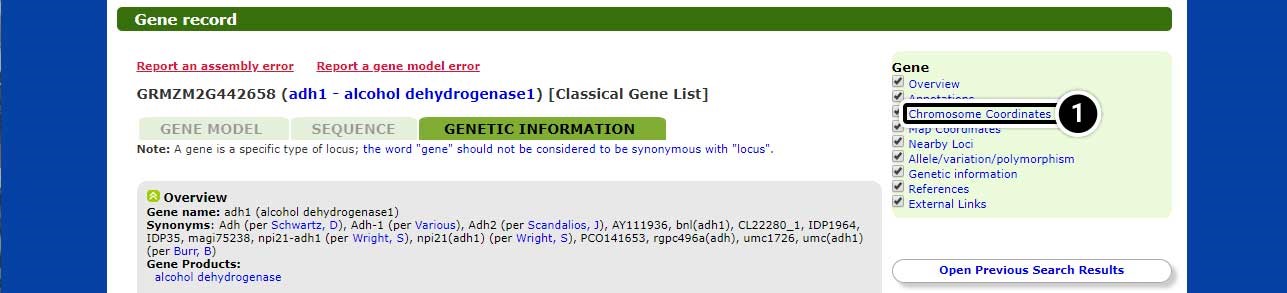
Step 4: Explore the Gene Record
The locus record screen provides detailed information on the adh1 gene. Explore the genetic information for adh1 alcohol dehydrogenase1.
- Click on Chromosome Coordinates when ready to proceed.
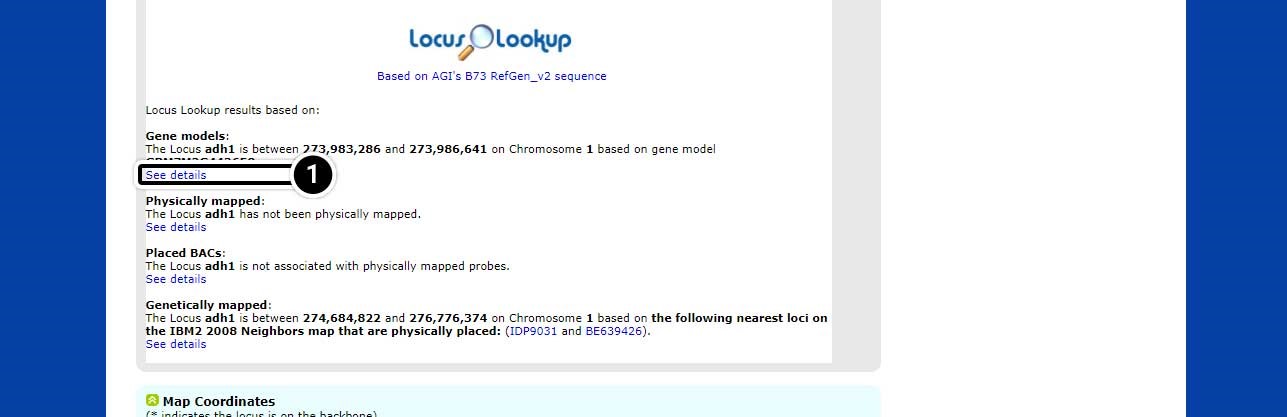
Step 5: See Details in Locus Lookup
The page will scroll down to the Locus Lookup section.
- Click on Show details to expand this section of the results.
Step 6: Expanded Details in Locus Lookup
When the details have loaded, explore the available information.
Note the position of adh1 based on “AGIs B73 RefGen_v2 sequence” (adh1 is located between 273,983,286 and position 273,986,641 on chromosome 1.
- Click on the map image to launch the MaizeGDB genome browser.
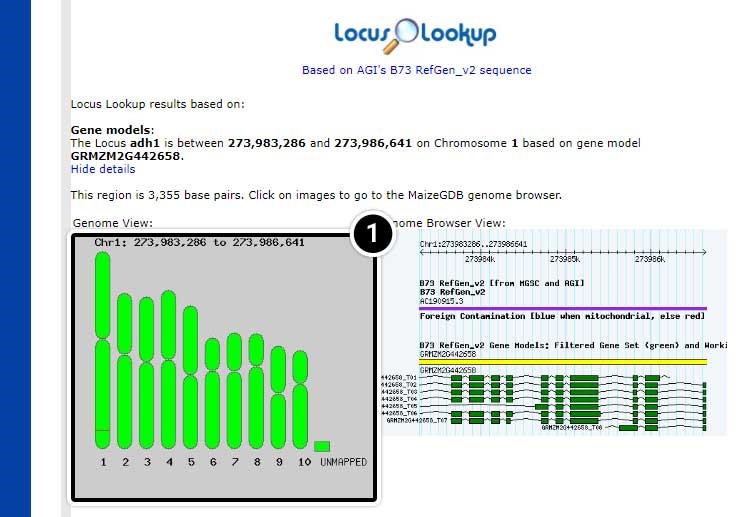
Step 7: View Datasets in the MaizeGDB Genome Browser
The MaizeGDB Genome Browser is displayed. Click the image below to see a larger version.
- Here you can use the other datasets available in MaizeGDB including B73 RefGen_v1 sequence, B73 RefGen_v3 sequence, B73 RefGen_v4 sequence, and BAC-based genome assembly.
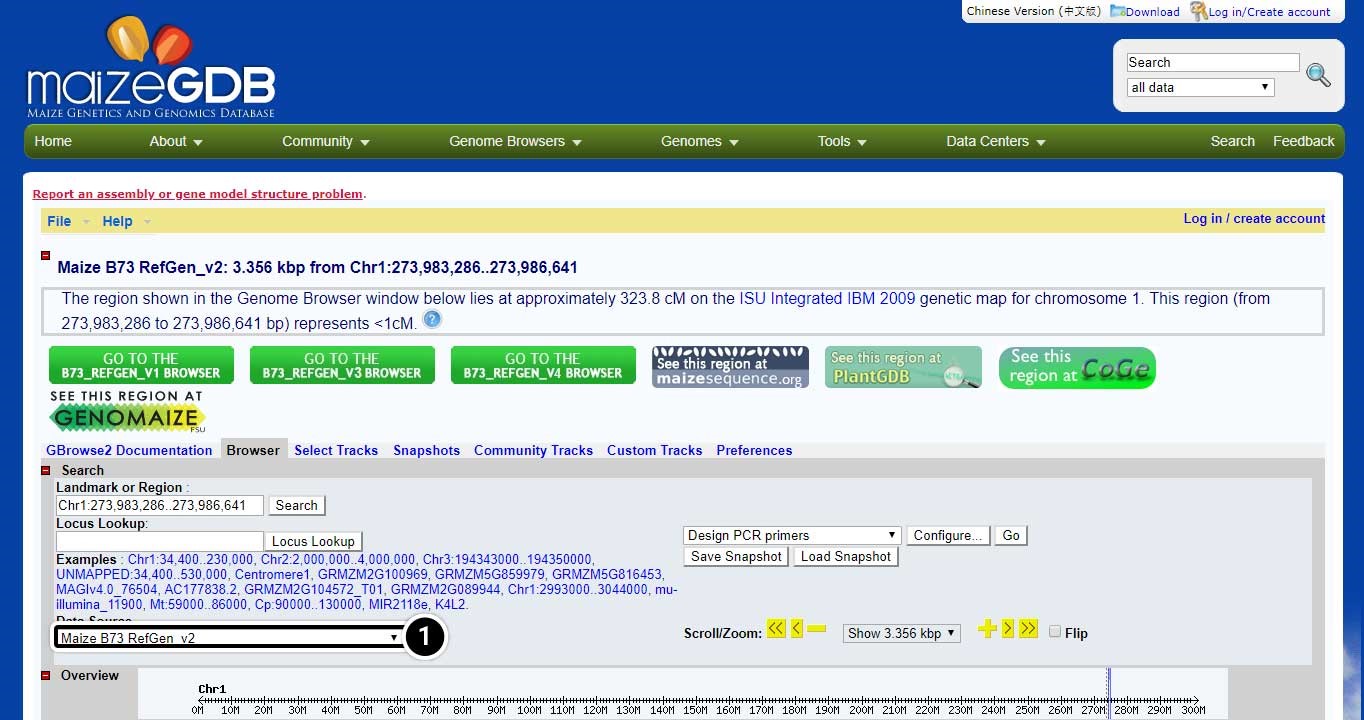
Next, we will conduct a BLAST search for adh1 in maize GDB using adh1 mRNA from GenBank.
- In the navigation bar, hover over Tools
- Then click the BLAST button.
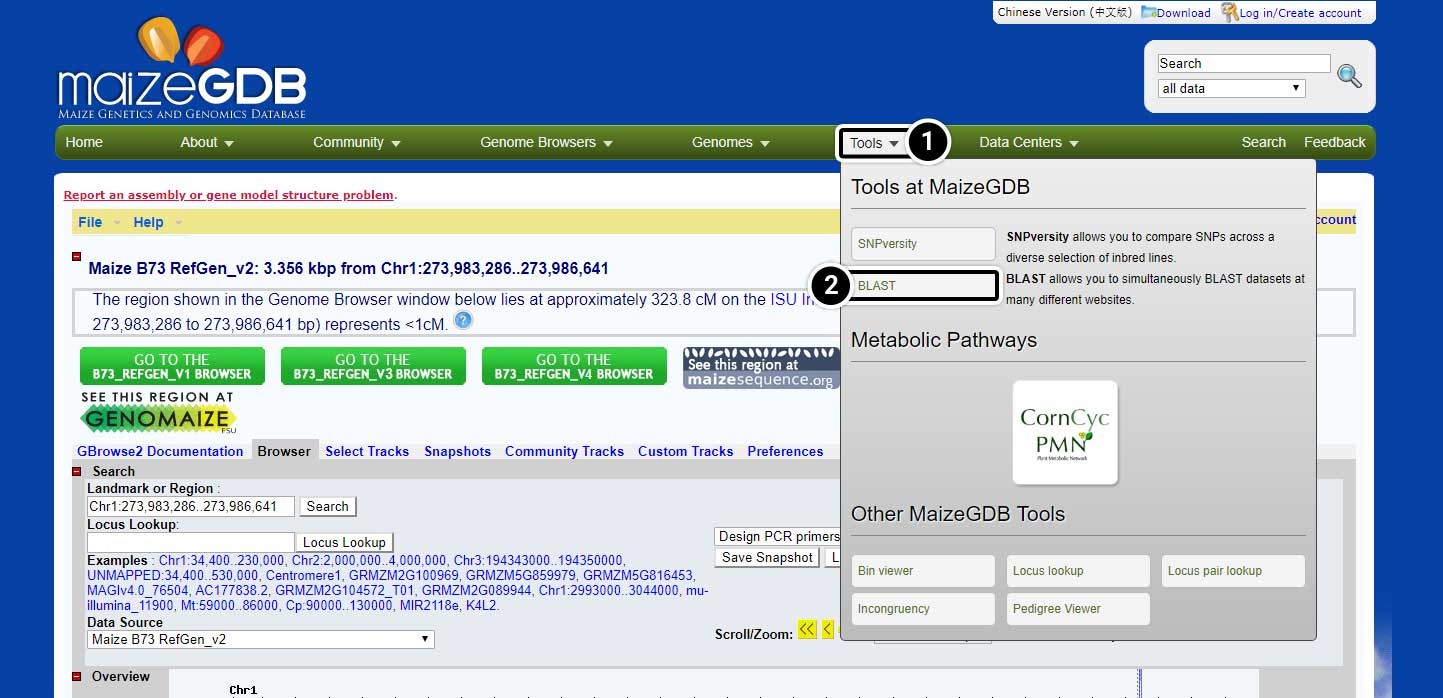
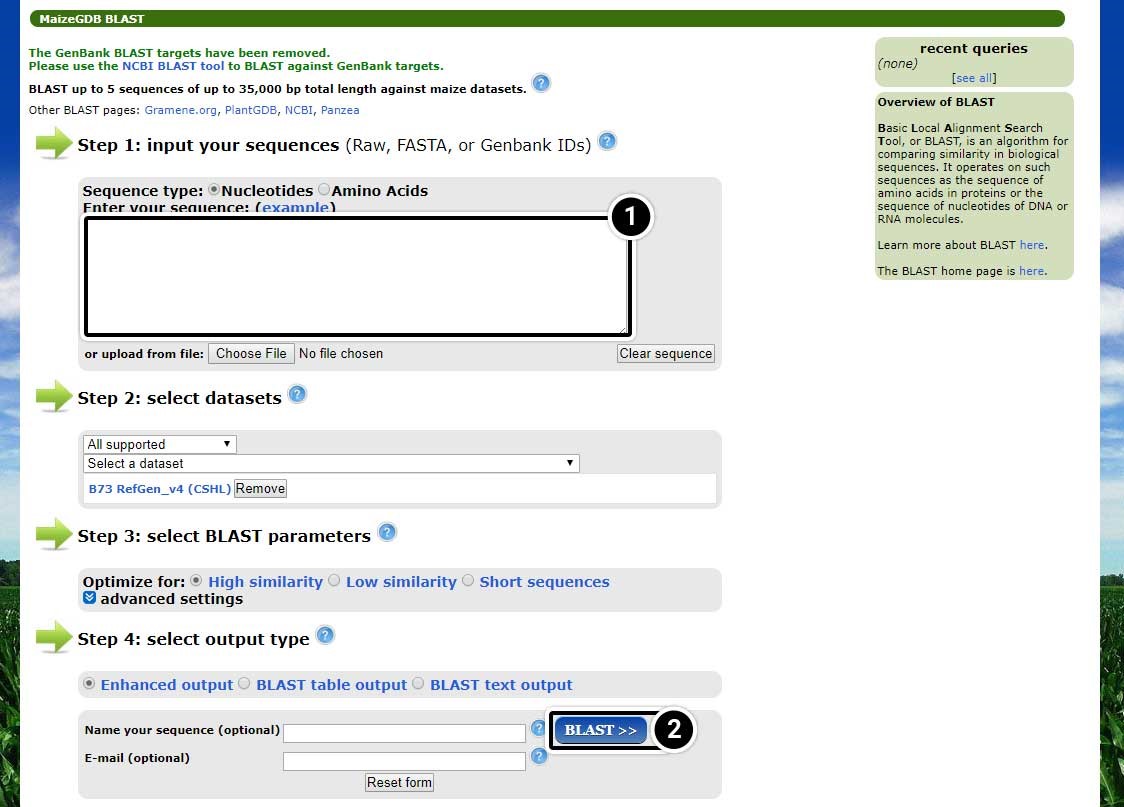
Input the BLAST Parameters
- Enter the adh1 mRNA sequence in FASTA format in the box.
- Use the default parameters to search for adh1 and click the BLAST button.
View the BLAST Results
The table of BLAST results includes information on chromosomes, probability values, sequence identity, and the number of likely candidates (hits). Also, you can view a representation of the entire genome in the context of where adh1 may be located.
- Click the arrow next to “Whole genome view” to see the entire genome in context of where adh1 may be located.
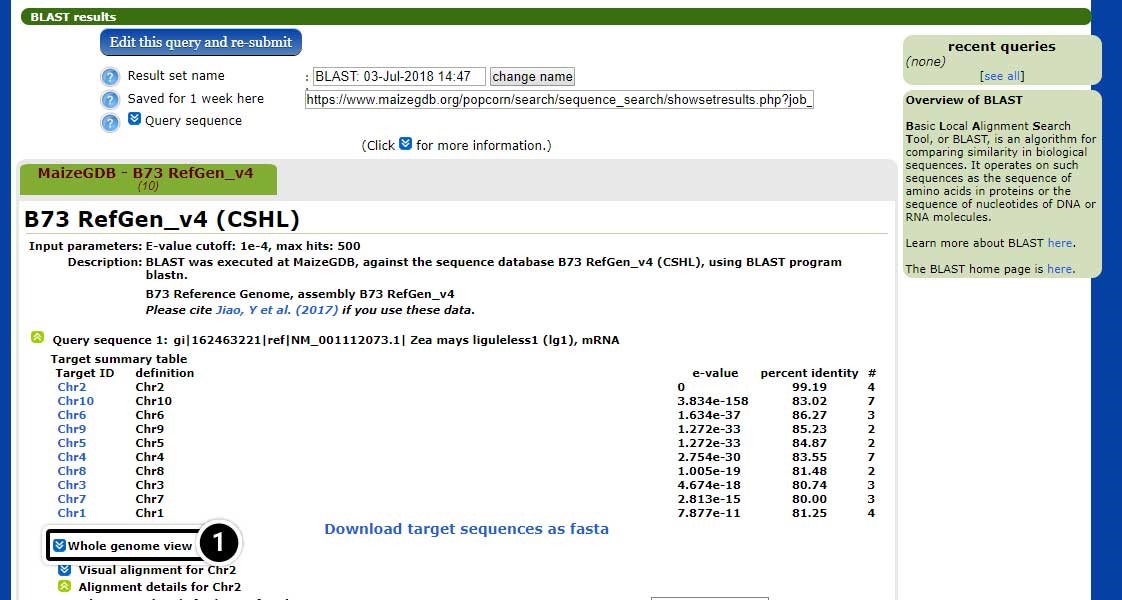
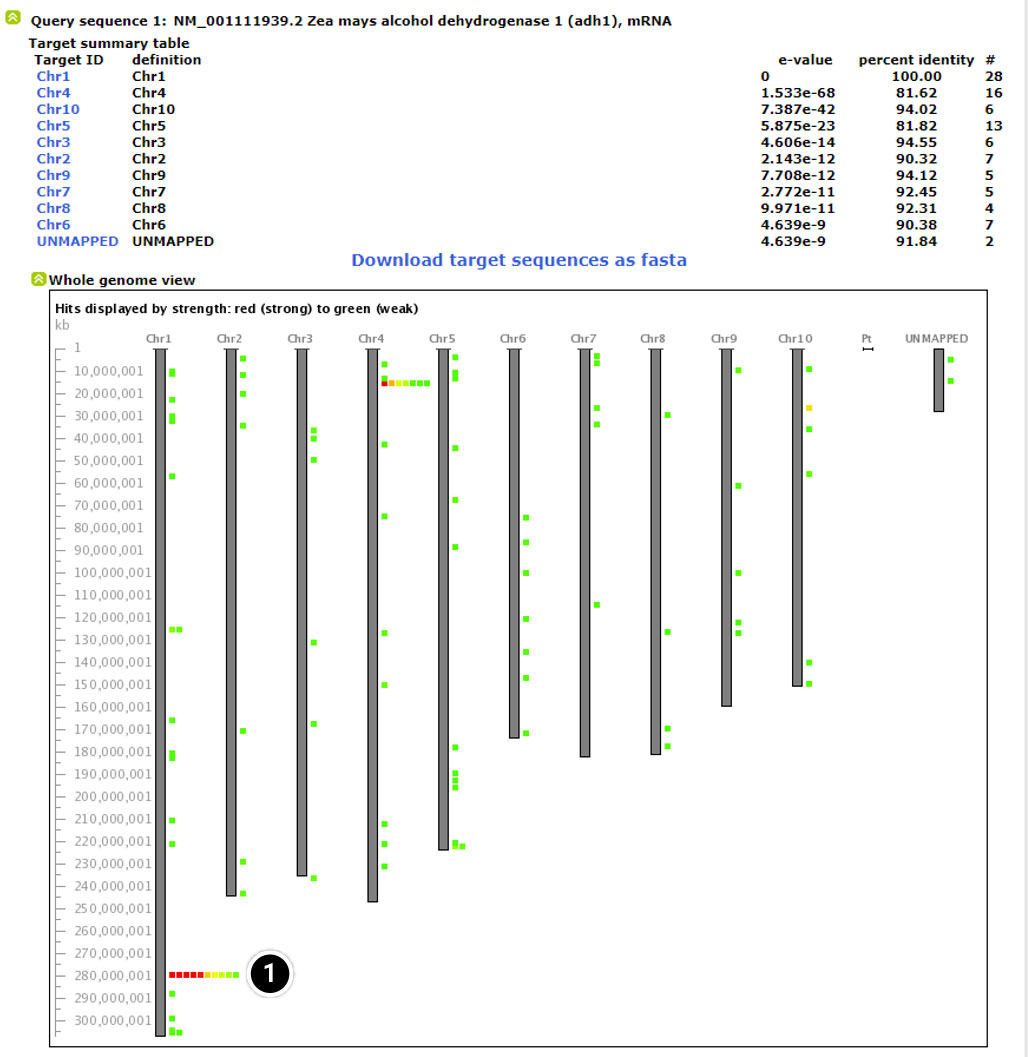
Explore the Whole Genome View
The whole genome view allows visualization of the 10 chromosomes of maize including, the predicted position match the adh1 sequence.
- Click on “Chr1” corresponding to the red box on Chromosome 1 (E-value = 0).
- Now, click on “View at MaizeGDB”, next to the hit on Chr1.

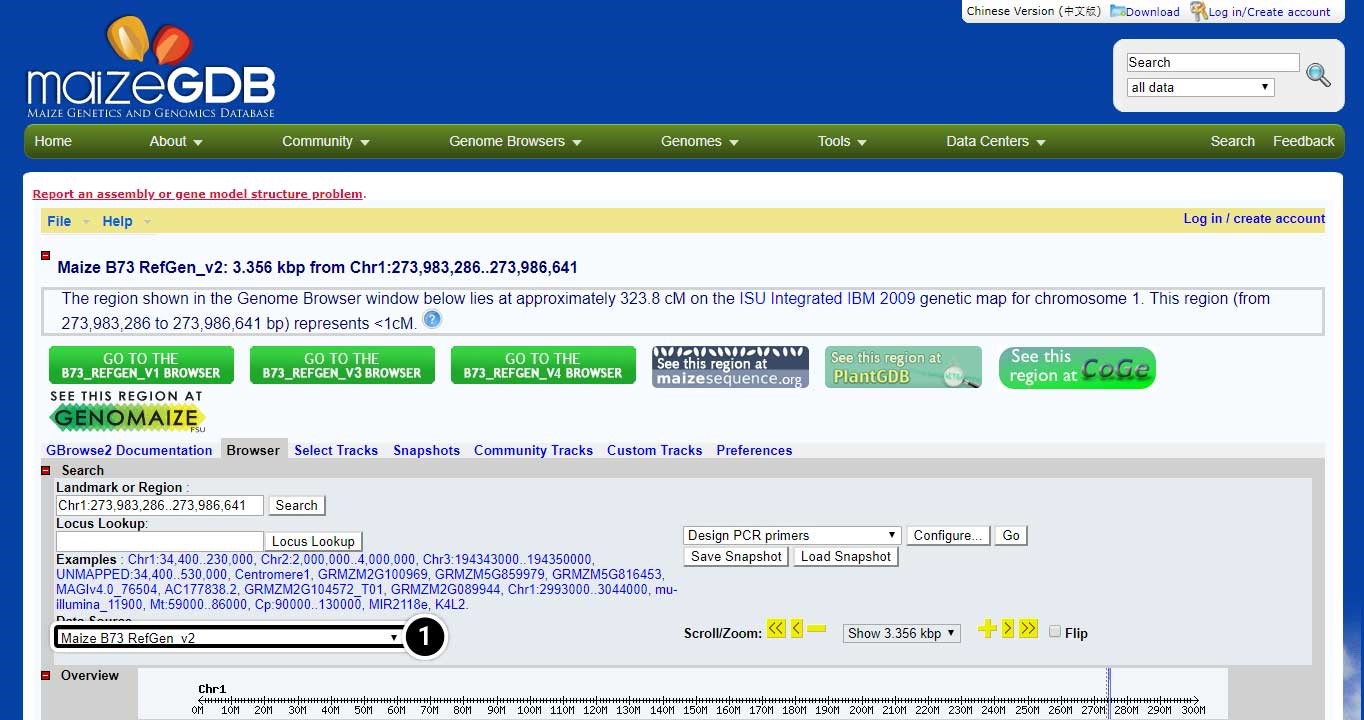
Change the Data Source
The selection you made on the last screen will open a new window containing information on the position of adh1 and data sources.
- Click on the pull-down menu of “data source” (arrow) to explore other data sets.
Possible Explanation for BLAST Study Question Results
One reason for discrepancies might be that there are in this genomic region several copies of the gene (eventually ancient duplication no longer actively transcribed due to mutations or whatever). Depending on the origin of the query sequence you use to find the gene, they might show different hit scores from these versions of the gene. As for the version2 pseudo-molecule, the location seems to be quite similar…
Multiple Sequence Alignment
Some of the key steps in building a multiple alignment include:
- Obtain the sequences to align by database searching
- Run the multiple alignment program and,
- Identify the residues that differ or are conserved among the sequences (finding polymorphisms)
Enter the NCBI site and use the following steps to guide your activity.
Search the NCBI Website for the Allelic Sequences
Find the allelic sequences for a maize gene. Here we will use teosinte branched1 (tb1) gene from maize as an example.
- Open your web browser and go to https://www.ncbi.nlm.nih.gov/.
- Enter tb1 AND Zea[orgn] into the search box.
- Press Enter or click Search.
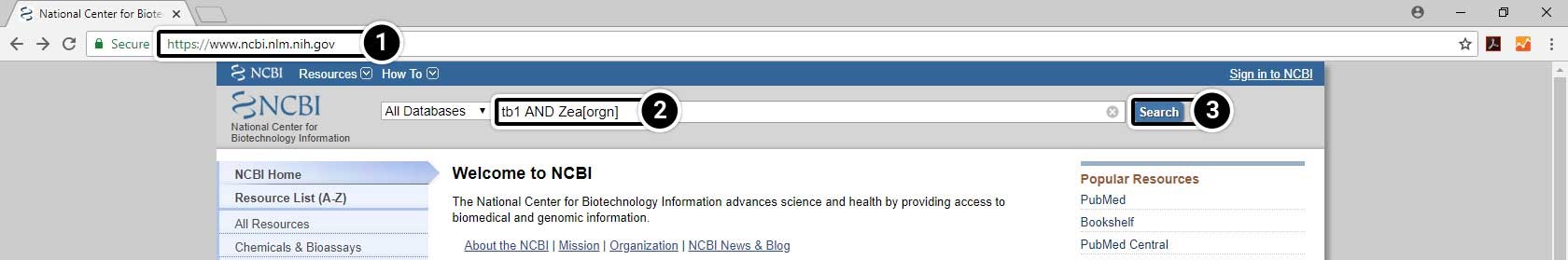
Narrow the Search Results
- Select PopSet (population data sets).
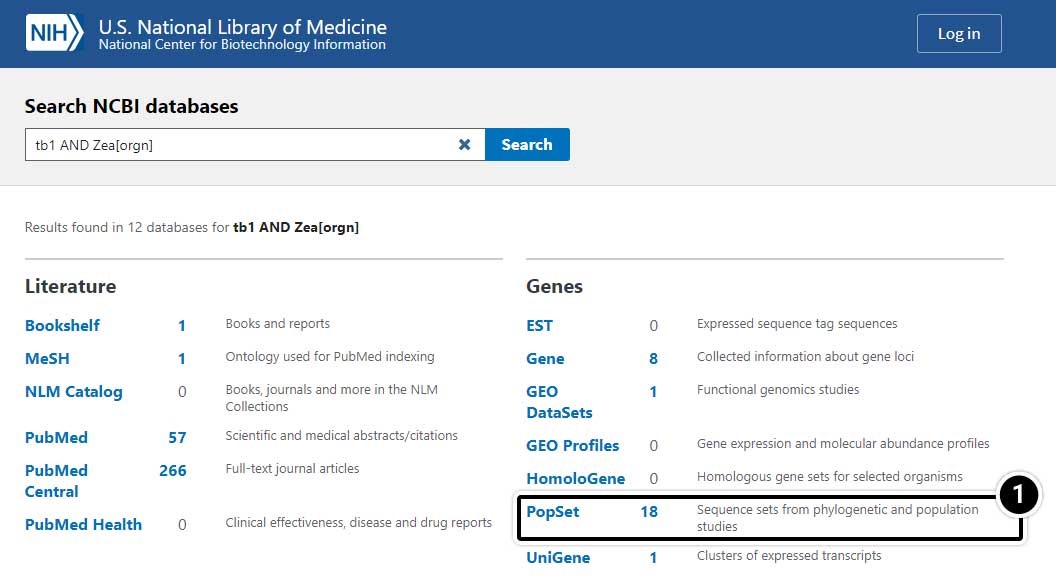
Choose the Specific Search Result
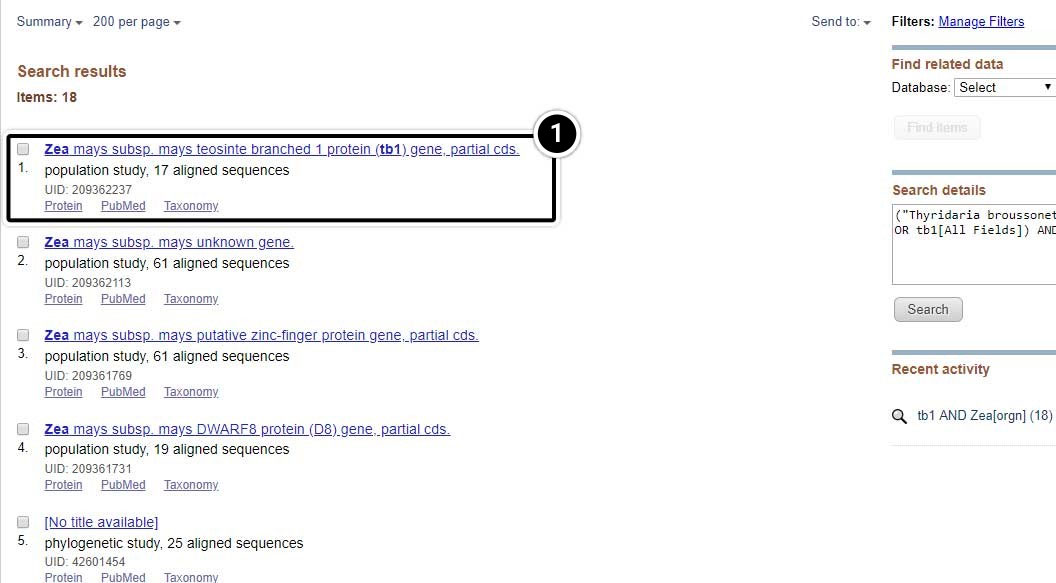
- Select the result containing 17 aligned sequences of tb1 partial cds from a population study. (UID 209362237)
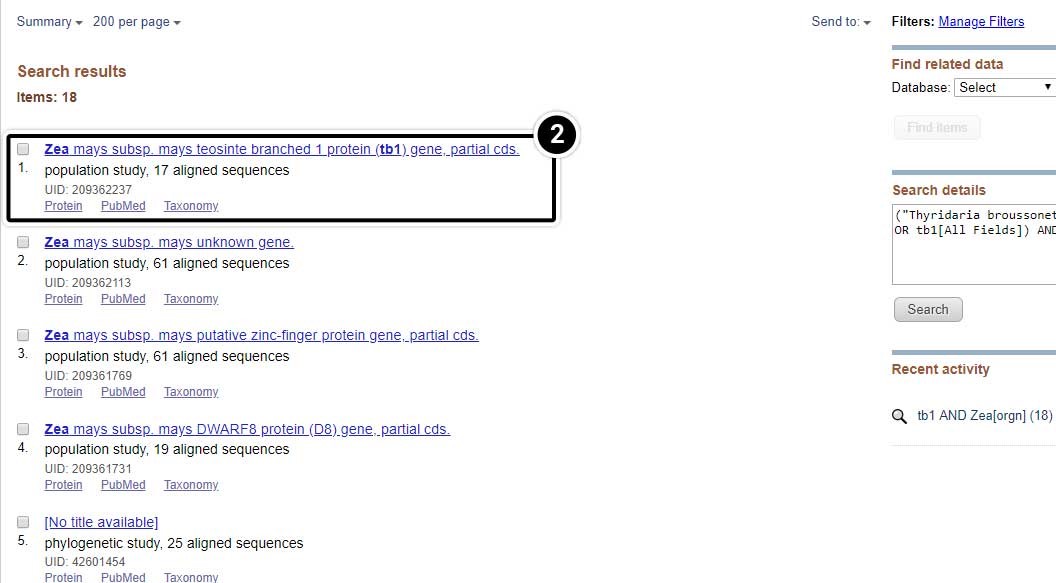
Explore the Alignment
Scroll down to see the alignment of the 17 tb1 partial cds.
- Click the + sign until you can see the nucleotides.
- Click the arrow to pan right in the sequences until you can see the region between 1500 and 1530.
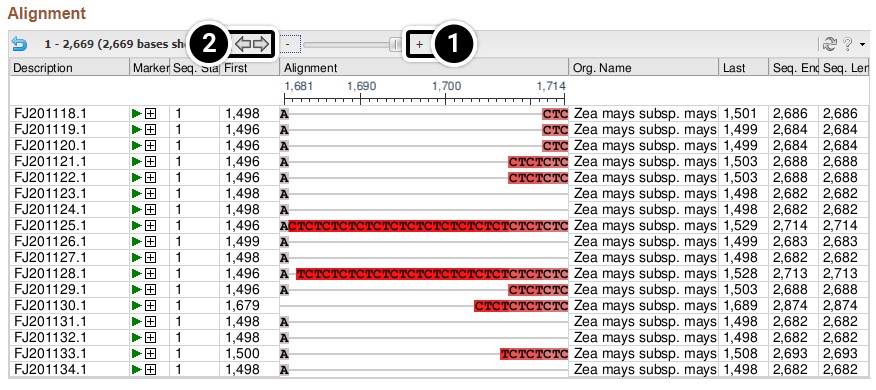
Review your output from this activity.
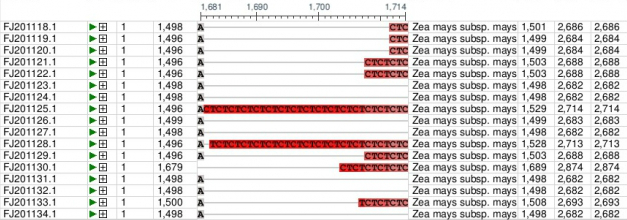
Finding Polymorphisms
Using Clustal Omega
To detect polymorphisms in a set of candidate genes requires a program that aligns multiple sequences. Clustal Omega is one of the commonly used programs. Clustal Omega is a hierarchical multiple alignment program that combines a robust method for multiple sequence alignment with a user-friendly interface. There are different webservers that provide access to Clustal Omega. For this lesson we will use the European Bioinformatics Institute webserver. Clustal Omega can also be downloaded to a personal computer for more routine use. The following is an example of how to use Clustal Omega to detect polymorphisms.
Search the NCBI Website
- Go to the NCBI website and search for tb1 AND Zea[orgn].
- Click Search
Explore the Search Results
- Select PopSet (population data sets).
Select the Population Set
- Click on the population set we studied earlier (UID 209362237)
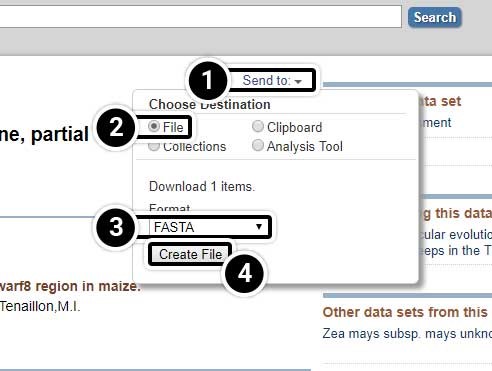
Create a FASTA File
Create a FASTA file of the 17 tb1 sequences.
- Click the pull-down menu Send to: at the top right of the screen
- In the menu that appears, select File for the destination
- Select the FASTA format
- Finally, click Create File.
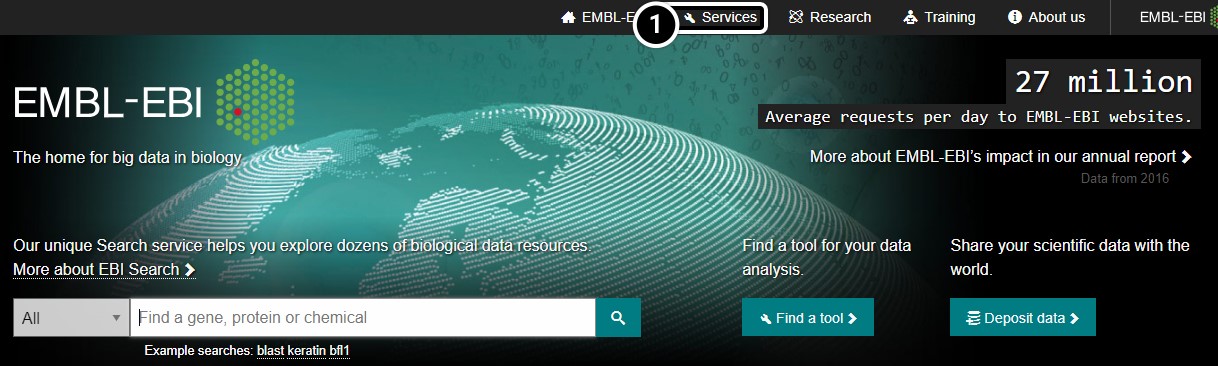
Access Clustal Omega
Access the Clustal Omega program through EMBL-EBI.
- Click the Services link
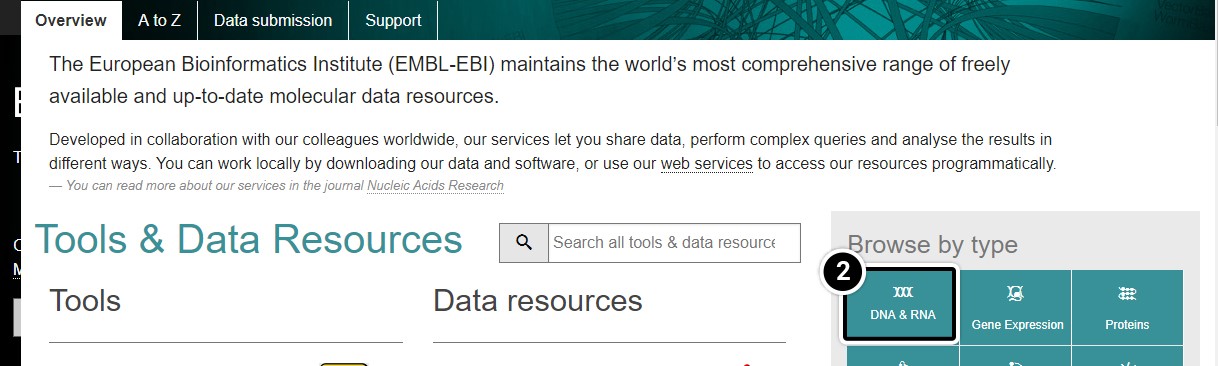
- Under Browse by type, click DNA & RNA
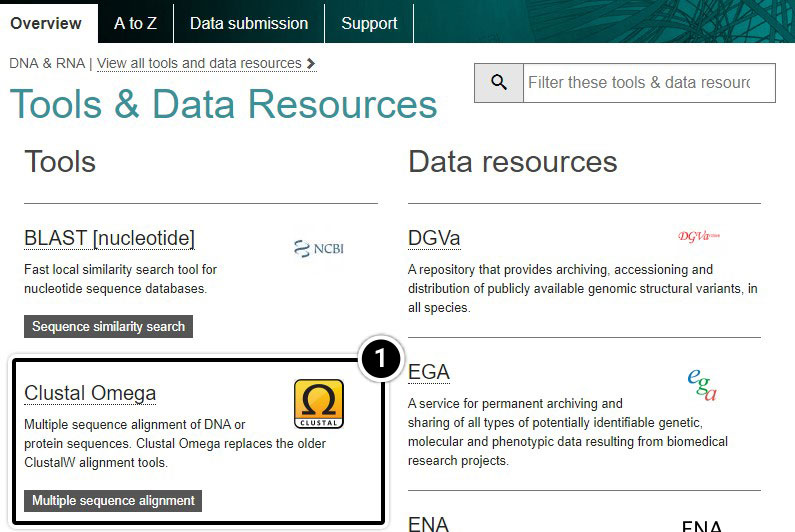
Perform Alignment
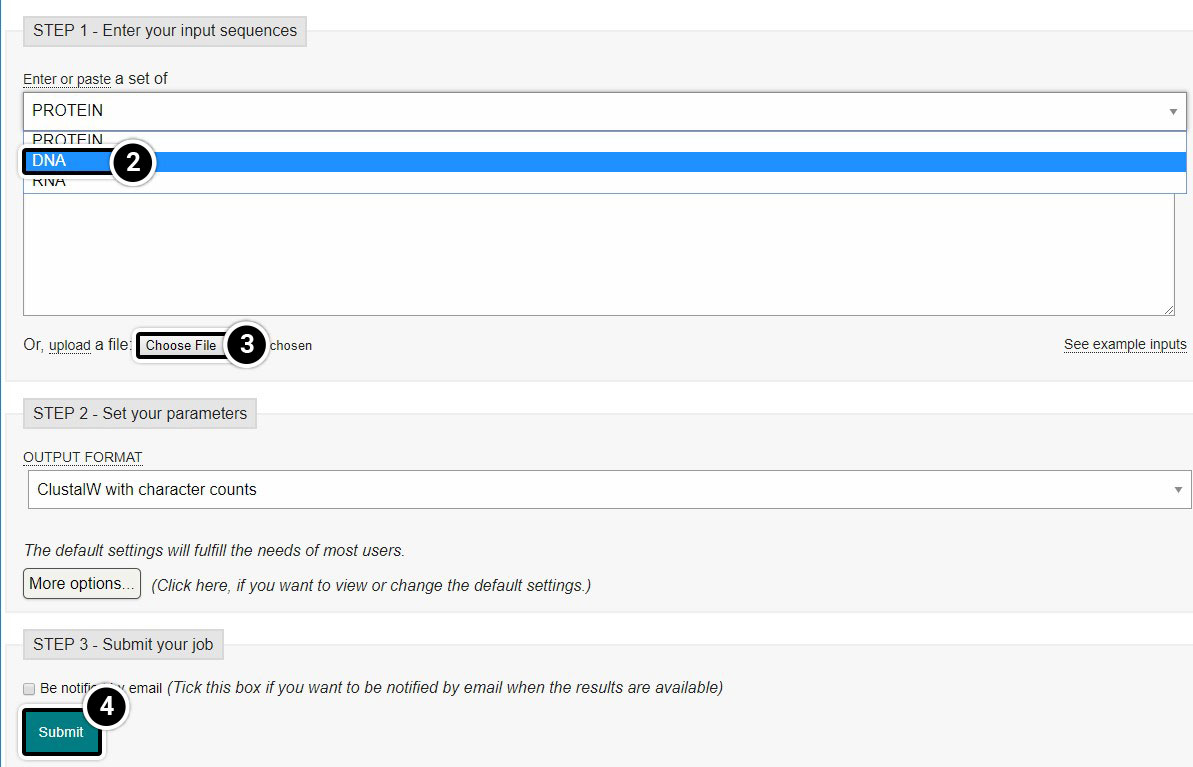
Perform alignment of tb1 partial cds using Clustal Omega. Within the Clustal Omega window you have the option of pasting sequences, or uploading files containing your sequences in FASTA format. We will upload the FASTA file you created in Step 2. As you may notice in this window, the default is set as “PROTEIN.” Since you wish to align tb1 DNA sequences, you must change this parameter. Upload your file and click Submit.
- Click Clustal Omega
- Select DNA from the dropdown
- Click Choose File to browse for the file you created.
- Click Submit
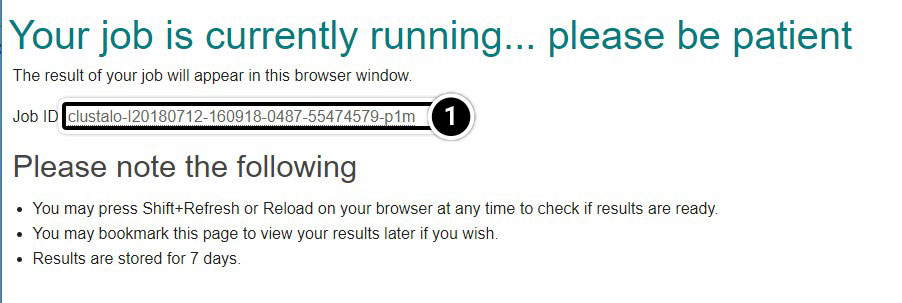
Explore the Output
It will take a moment before you obtain a report of your job request. You can click and save the “Your Job Output” URL to view your results for up to seven days.
- Click the Job ID link
- You can click the Download Alignment File but that is not necessary for this activity
- Click Result Summary
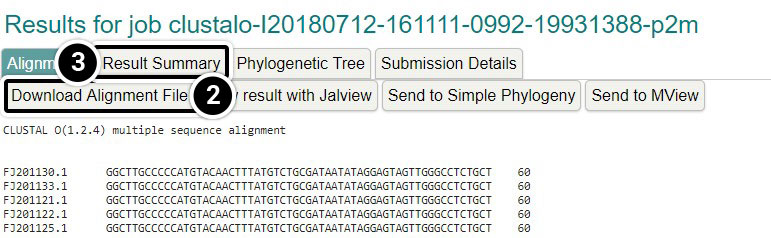
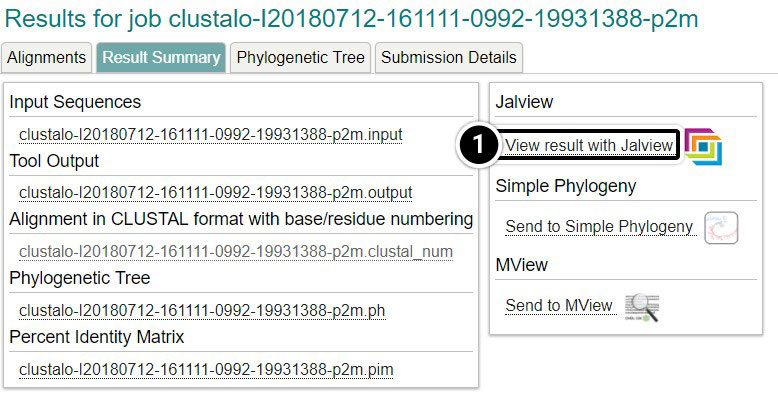
View Result with Jalview
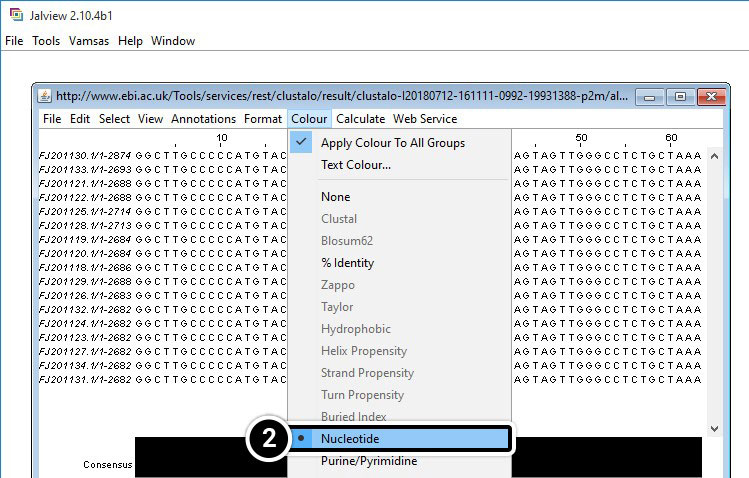
- Click View result with Jalview
- Once Jalview opens, click Colour then Nucleotide
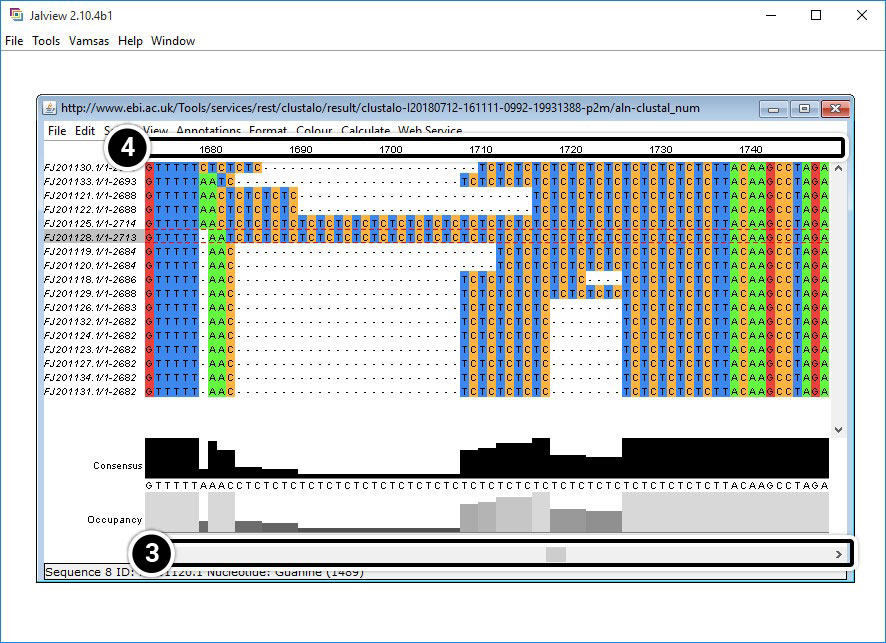
- Use the scroll bar to navigate to the alignment.
- Scroll to align the region from 1680 to 1740.
Compare Results from JalView and NCBI BLAST
Analyze region 1680 to 1740 of your JalView results (below).
What is unique about this region?
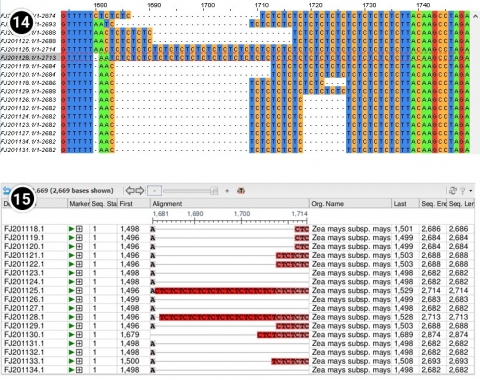
How does it compare with the region between positions 1500 and 1530 in the NCBI BLAST?
- JalView
- NCBI BLAST
Developing Marker Assays
Recall in Module 2 you learned how SSR and SNP can be analyzed by PCR and restriction enzymes. In lesson 8 of this course, you will learn additional strategies to detect DNA polymorphisms for marker development.
Summary
Biological sequence databases serve an important role of providing access to sequence information to the research community. Searches can be restricted to a single database or expanded to include all other databases. Whole genomes can be explored to predict positions that match a specific sequence. To detect polymorphisms in a set of candidate genes a program that aligns multiple sequences is required. The detected polymorphisms can be used to develop markers to assist in selection.
